
Welcome to the
Malaysian Family Physician, a peer-reviewed open-access journal of family practice
and primary care research.
ORIGINAL ARTICLE
THE BLUE MAN WHO PRESENTED WITH A STROKE
KC Koh MMed (Internal Medicine), MBBS (Manipal); MH Wong MMed (Internal Medicine), MBBS (IMU)
Department of Medicine, Clinical School, International Medical University, Seremban, Negeri Sembilan, Malaysia (Koh Kwee Choy, Wong Mun Hoe)
Address for correspondence: Dr Koh Kwee Choy, Senior Lecturer, Department of Internal Medicine, Clinical School, International Medical University, Jalan Rasah, 70300 Seremban, Negeri Sembilan, Malaysia. Tel: +606-7679 896, Fax: +606-7677 709. Email: kweechoy_koh@imu.edu.my
A 19-year-old man presented with sudden onset of right eye ptosis, diplopia and giddiness. He had no previous medical illnesses with negative history of exertional dyspnoea, epistaxis, haemoptysis, palpitations, chest pain and chronic cough. Examination revealed central cyanosis, digital clubbing, polycythaemia, partial ptosis of right eye, diplopia on right gaze and dilated right pupil. Examination of the chest revealed pectus excavatum but no cardiac murmurs were heard. Investigations revealed a solitary right pulmonary arteriovenous malformation with two feeder vessels which were successfully embolized surgically.
Keywords: Cyanosis, clubbing, pulmonary arteriovenous malformation.
Koh KC, Wong MH. The blue man who presented with a stroke. Malaysian Family Physician. 2011;6(1):29-31
INTRODUCTION
Pulmonary arteriovenous malformations (PAVM) are caused by abnormal communications between pulmonary arteries and pulmonary veins, which are commonly congenital (up to 80%) in nature.1,2 This condition is rare with an estimated incidence of 2-3 per 100,000 population.3 We report a case of solitary PAVM in an asymptomatic 19-year-old man who presented with right pontine infarct which was most likely due to paradoxical embolism across the PAVM.
CASE REPORT
A 19-year-old man presented with sudden onset of right eye ptosis, diplopia and giddiness. He had no previous medical illnesses and gave no history of exertional dyspnoea although he admitted to a sedentary lifestyle and avoided active physical activities by preference. He also did not have episodes of recurrent epistaxis, haemoptysis, palpitations, chest pain and chronic cough. Physical examination revealed central cyanosis (oxygen saturation on room air was 78-80%), digital clubbing, polycythaemia, partial ptosis of right eye, dilated right pupil with diplopia on right gaze due to right third cranial nerve palsy. On further questioning, he did not notice the bulbous nature of his fingers and toes nor did he notice the bluish tinge of the nail beds. Examination of the chest revealed pectus excavatum. No cardiac murmurs or pulmonary bruit were heard. The patient had no physical examination evidence of Osler-Weber-Rendu Syndrome.
Complete blood count revealed polycythaemia (Hb of 210 g/L; HCT 0.64 L/L). Computed Tomography (CT) brain showed evidence of a right pontine infarct with no suggestion of cerebral arteriovenous malformation. Chest radiograph showed a faint right mid-zone opacity. Transthoracic echocardiography showed no evidence of septal wall defects or large vessel anomalies and no pulmonary hypertension. Plain CT of the chest suggested the presence of a PAVM (Figure 1). This was confirmed by CT pulmonary angiogram which revealed a solitary PAVM with two feeder vessels in the lower lobe of the right lung (Figure 2 and 3).
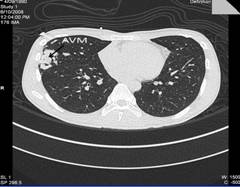
Figure 1: CT scan of the chest suggested the presence of right PAVM.
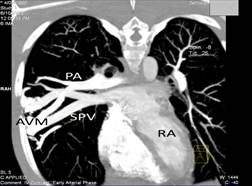
Figure 2: 2-D reconstruction of CT pulmonary angiogram in early arterial phase reveals the presence of a solitary right PAVM. PA: pulmonary artery; SPV: superior pulmonary vein; AVM: arteriovenous malformation; RA: right atrium.
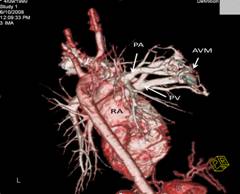
Figure 3: 3-D reconstruction of CT pulmonary angiogram shows in greater detail the AVM with feeder vessels from the right pulmonary artery (PA). PV: pulmonary vein; AVM: arteriovenous malformation; RA: right atrium.
Under general anaesthesia, the feeders were subsequently occluded with an Amplatzer Vascular plug size 8 x 7 mm and a 5 x 5 mm Gianturco Cook coil, respectively. The patient made an uneventful recovery with post-procedure oxygen saturation at 100% on room air and complete resolution of central cyanosis.
DISCUSSION
The age at first presentation of PAVM ranges from newborn to 70 years but most cases are diagnosed in the first three decades of life.2,4,5 PAVM occur twice as often in females although there is a male predominance in newborns.6 The anomaly may be solitary or multiple where the incidence of the former ranges from 42-74% and are found in the lower lobes of the lung.7 Approximately 70% of PAVM are associated with hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu Syndrome, which is associated with cerebral arteriovenous malformations (AVM).1
The most common presenting symptom is dyspnoea on exertion, although 13 to 55% of patients may be asymptomatic. This patient did not have exertional dyspnoea. Most patients with PAVM tolerate hypoxaemia very well unless arterial oxygen level falls below 60 mmHg.2,7,8 In a case series report of 126 patients with PAVM, Cottin et al.9 reported that while 43% of the patients were hypoxemic at rest, only 22% had dyspnoea. The absence of exertinal dyspnoea in this patient may have contributed to the delay in the diagnosis of his condition. Other symptoms include recurrent episodes of epistaxis, haemoptysis, palpitations, chest pain and cough.7 The commonest physical findings are cyanosis, clubbing, and pulmonary vascular bruit.10,11 Polycythaemia is common although patients with HHT may have telangiectasia and anaemia. The presence of anaemia in patients with HHT may sometimes mask cyanosis.1,7
In the case series report by Cottin et al.,9 only 56% of PAVM are visible on plain chest radiograph. CT pulmonary angiography remain the gold standard for confirming the presence of PAVM especially when plain CT or Magnetic Resonance Imaging (MRI) are inconclusive. Other useful but less accessible diagnostic tools include shunt fraction measurement, contrast echocardiography and radionuclide perfusion lung scanning. Family members of patients who fulfil the criteria for diagnosis of HHT (namely, recurrent and spontaneous epistaxis, multiple mucocutaneous telangiectases, visceral lesions e.g. gastrointestinal or pulmonary AVM, and first-degree relative with HHT)12 should be screened for possible PAVM since there is a high incidence of unsuspected PAVM in this population.
Untreated PAVM are associated with considerable morbidity and mortality where approximately 25% of PAVM gradually enlarge over time2 leading to potentially fatal complications such as stroke, cerebral abscess, hemoptysis due to intrabronchial rupture, or hemothorax due to intrapleural rupture. Other complications include progression to development of pulmonary hypertension, endocarditis, migraine headaches, congestive heart failure, and transient ischaemic attack (TIA).7
The risk of neurologic complications increases with age and number of PAVM. Moussouttas et al.13 in a case study of 75 cases of PAVM, reported a strong association between single PAVM and various neurologic manifestations with greater prevalence for patients with multiple PAVM, a detailed study of neurologic complications in 71 patients with HHT and PAVM by Maher et al.14 found that embolic neurologic complications (stroke, TIA, or cerebral abscess) occurred in 10% of patients less than 30 years old and in 45% of older patients. White et al.15 reported the incidence of neurological events associated with PAVM to be as follows: migraine (43%), TIA (37%), CVA (18%), abscess (9%), and seizure (8%). This patient suffered a pontine infarct and the cause was most likely due to paradoxical embolism across the PAVM.13
The current preferred treatment for PAVM is percutaneous embolotherapy using coils or balloons. Surgical resection of PAVM is indicated in patients who fail embolotherapy, develop serious bleeding complications despite embolotherapy, have intrapleural ruture of the PAVM or are unsuitable for embolotherapy due to allergy to contrast.7
CONCLUSION
PAVM are rare and often described as an ‘orphaned’ condition. The classical triad of clubbing, cyanosis and dyspnoea should prompt the clinician to consider its possibility. Untreated, PAVM is associated with considerable morbidity and mortality. It is also strongly associated with HHT and patients with PAVM should be screened for presence of cerebral AVM. Contrast CT of the chest or pulmonary angiography is usually diagnostic of PAVM although the latter remains the gold standard. Embolotherapy is the preferred safe and non-invasive management strategy for PAVM.
The PAVM in this patient was unfortunately diagnosed only after he suffered a right pontine infarct which was most likely secondary to paradoxical embolism across the PAVM. Although he had no dyspnoea, he did have gross clubbing and cyanosis. A negative echocardiogram which ruled out congenital heart disease prompted further investigations which confirmed the diagnosis of a solitary PAVM on CT pulmonary angiogram which was successfully embolized.
ACKNOWLEDGEMENT
We acknowledge Dr Geetha A/P Kandavello, National Heart Institute, Malaysia for her contribution during the embolotherapy.
REFERENCES
- Gossage JR, Kanj G. Pulmonary arteriovenous malformations. A state of the art review. Am J Respir Crit Care Med. 1998;158(2):643-61. [PubMed] [Full text]
- Dines DE, Arms RA, Bernatz PE, et al. Pulmonary arteriovenous fistulas. Mayo Clin Proc. 1974;49(7):460-5. [PubMed]
- Hodgson CH, Kaye RL. Pulmonary arteriovenous fistula and hereditary hemorrhagic telangiectasia: a review and report of 35 cases of fistula. Dis Chest. 1963;43:449-55. [PubMed] [Full text]
- Prager RL, Laws KH, Bender HW Jr. Arteriovenous fistula of the lung. Ann Thorac Surg. 1983;36(2):231-9. [PubMed]
- Sharifah AI, Jasvinder K, Rus AA. Pulmonary arteriovenous malformation: a rare cause of cyanosis in a child. Singapore Med J. 2009;50(4):e127-9. [PubMed] [Full text]
- Allen SW, Whitfield JM, Clarke DR, et al. Pulmonary arteriovenous malformation in the newborn: a familial case. Pediatr Cardiol. 1993;14(1):58-61. [PubMed]
- Khurshid I, Downie GH. Pulmonary arteriovenous malformation. Postgrad Med J. 2002;78(918):191-7. [PubMed] [Full text]
- Dines DE, Seward JB, Bernatz PE. Pulmonary arteriovenous fistulas. Mayo Clin Proc. 1983;58(3):176-81. [PubMed]
- Cottin V, Chinet T, Lavole A, et al. Pulmonary arteriovenous malformations in hereditary hemorrhagic telangiectasia: a series of 126 patients. Medicine (Baltimore). 2007;86(1):1-17. [PubMed]
- Stringer CJ, Stanley AL, Bates RC, et al. Pulmonary arteriovenous fistula. Am J Surg. 1955;89(5):1054-80.[PubMed]
- Sluiter-Eringa H, Orie NG, Sluiter HJ. Pulmonary arteriovenous fistula. Diagnosis and prognosis in noncomplainant patients. Am Rev Respir Dis. 1969;100(2):177-88. [PubMed]
- Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet. 2000;91(1):66-7. [PubMed]
- Moussouttas M, Fayad P, Rosenblatt M, et al. Pulmonary arteriovenous malformations: cerebral ischemia and neurologic manifestations. Neurology. 2000;55(7):959-64. [PubMed]
- Maher CO, Piepgras DG, Brown RD Jr, et al. Cerebrovascular manifestations in 321 cases of hereditary hemorrhagic telangiectasia. Stroke. 2001;32(4):877-82. [PubMed] [Full text]
- White RI Jr, Lynch-Nyhan A, Terry P, et al. Pulmonary arteriovenous malformations: techniques and long-term outcome of embolotherapy. Radiology. 1988;169(3):663-9. [PubMed]